
“Toxicokinetics is basically the study of “how and what happens to a substance in the body”
Table of Contents
What is Toxicokinetics?
Toxicokinetics (TK) is characterised as the generation of pharmacokinetic ( PK) data, either as an integral component in conducting non-clinical toxicity studies or in supporting studies specifically designed to assess systemic exposure.
TK explains the use of bioanalytical sampling as a way of characterising the disposition of a target compound in animal toxicity studies over time.
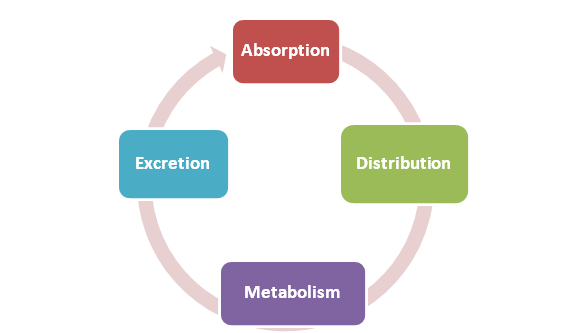
TK is closely related to PK which is defined in a living organism as the process of drug absorption, distribution, metabolism , and excretion (ADME).
In order to obtain an understanding of their effects, all drugs under development for the treatment of human diseases must undergo a series of PK / TK investigations in animals and humans. The primary objective of toxicokinetics is to explain the systemic exposure obtained in animals and its relation to the dosage level and the time course of the toxicity research.
TK represents the ADME of a molecule in large measure as it passes around an organism’s body.
As for all animal research, variations in interspecies in ADME should be considered before clinical human testing and during extrapolation in results.
TK studies may have quantifiable endpoints if adequately used to help illustrate the harmful effects that can occur during toxicity studies.
Toxicokinetics versus Pharmacokinetics
While TK shares important parameters with preclinical PK, such as Cmax and AUC, the studies are distinct in several ways:
TK’s primary objective is to associate toxicity results (not therapeutic effectiveness) with a corresponding degree of exposure to an experimental drug compound.
TK experiments also use doses that are much greater than the therapeutically appropriate doses would be.
Administering these higher doses could potentially yield distinct kinetics from those of PK trials, which could inform both non-clinical and clinical dosing considerations and drug safety margins in later stages of drug development.
In general, the TK arm of a non-clinical toxicology study has fewer time points, fewer subjects and fewer endpoints than non-clinical and clinical PK studies.
Half-life (t1⁄2) may not be accurately determined in TK studies (though frequently estimated) due to relatively sparse concentration-time sampling of blood or plasma.
How Does ADME Influence Toxicokinetics?
In animals as well as humans, xenobiotics, or foreign chemical compounds, during their passage through the body, may experience drastic changes in location and chemical composition. In preclinical studies promoters expect to obtain as much specific knowledge as possible about their compound.
ADME directly impacts TK studies quantitatively, by impacting concentration versus time-dependent parameters, and qualitatively by generating molecular-level therapeutic effect and/or toxicity. Understanding the effects of ADME on TK will help sponsors get the biochemical puzzle pieced together.
Absorption
Absorption is the process in which a drug is absorbed by tissues in the body. In TK studies, how easily and where a drug is absorbed in the body may influence the route of administration. For example, drugs administered orally can take some time to be absorbed by the digestive tract and cross into systemic circulation via the gut-blood barrier. Drugs administered intravenously, on the other hand, move into the bloodstream immediately.
The route of administration and drug composition, which affects a drug compound’s rate of dissolution, may have a drastic effect on a specific pharmacological parameter: the bioavailability. Bioavailability is defined as the fraction of a dose given which enters the systemic circulation and may induce pharmacological effect.
To provide a more accurate picture of how a drug would be absorbed in later stages of development, it is possible to save time and money by selecting the best path and a formulation that closely imitates (and sticks to) the expected clinical formulation.
Distribution
Distribution defines where the drug goes until it enters the bloodstream and includes similar absorption factors.
In TK, distribution can be described as the reversible transfer of a molecule from the blood to other compartments, or the different body fluids that make up an organism.
At any given moment, the proportion of the total dose of the medication in the body to the drug concentration in the blood plasma provides a quantifiable, though theoretical, pharmacokinetic parameter: the volume of distribution.
Metabolism
Metabolism describes how the drug’s chemical composition changes as it moves across the body. We are especially concerned in drug metabolism with specific categories of enzymes which modify or add functional groups to the original drug molecule.
In TK, consideration of the effects of these chemical changes is particularly important, as certain compounds may still be active after one or more rounds of metabolism. The abundance of the expected metabolising enzymes in a TK experiment is also significant, as this can vary depending on the species, sex, type of tissue and age.
In selecting a suitable animal model for your study, consider whether the animal in response to drug administration develops similar metabolites as humans. If humans produce unique or significantly higher quantities of certain metabolites, it may be important to refining your TK strategy.
Excretion
Excretion is the process of an active drug compound which leaves the body with its metabolites. Each of the key parameters can affect excretion rate and path.
Rate of excretion issues during a TK study for overall exposure; slow excretion can exacerbate toxic effects, but rapid excretion can suggest a lack of distribution to target tissues.
In animal TK studies, like in humans, polar drug compounds may be voided intact by the kidneys through the urine. The liver can further modify parent drug compounds which contribute to bile and urine excretion.
Sampling and analysis of these materials can provide useful information on the percentage of the drug excreted in one route versus the other, and the identity of metabolites relevant to each route.
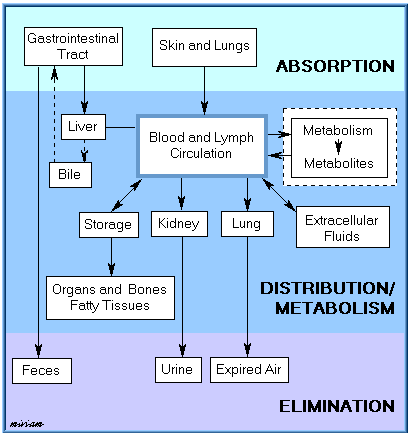
Absorption, distribution, biotransformation, and elimination are inter-related processes as illustrated in the following figure.
https://www.medicilon.com/what-is-toxicokinetics/
Microsampling in Toxicokinetics
Advances in microsampling technology and the development of more responsive analytical techniques have greatly enhanced the performance of TK studies over the last decade.
Microsampling is a technique that extracts 10–50 microliter (μL) of blood draws from laboratory animals at regular intervals after drug injection, depending on the sensitivity of the bioanalytical process.
These advances have generated several benefits for the field of TK, including:
- Smaller amounts of collection reduce the adverse effects of hemostasis, thereby reducing the impact on specific toxicological data.
- With less haemostasis impacts on major toxicity study animals, the need for TK satellite studies (studies conducted separately from general toxicity studies) is decreased and resources are saved.
- Rather than comparing satellite groups with main toxicity study animals, toxicity findings in an individual animal can be directly associated with TK in that animal.
- TK studies can be conducted with increased granularity, resulting in more precise endpoints.
In an updated 2018 guidance (S3A Guidance), FDA outlined the underlying benefits of using microsampling as it aligns with the field ‘s commitment to reduction, replacement and improvement (the 3Rs) by reducing the need for additional animals and larger blood samples. Your software will benefit from the cost savings of needless studies not running.
conclusion
In conclusion, TK studies share many common parameters with PK studies but the objective of these types of studies and their respective dose levels differ.
TK studies specifically serve an important role in the production of preclinical drugs as they link exposure (AUC and Cmax) to toxic effects from high doses of an experimental drug.”
https://journals.sagepub.com/doi/pdf/10.1177/019262339502300207